
Regulatory Requirements for the Validation of Analytical Methods – Part 1
Excerpt from the GMP Compliance Adviser, Chapter 14.D Validation of analytical test methods
7 min. reading time | by Joachim Ermer
Published in LOGFILE 03/2025
Analytical methods are used throughout the development, manufacture and release of pharmaceutical substances and dosage forms. The accuracy and reliability of analytical results are therefore essential. Successful method validation proves that the test procedure is suitable for the defined application. In today's feature, Joachim Ermer writes about the regulatory requirements for the validation of analytical methods.
In the next LOGFILE you will learn more about important aspects of putting these requirements into practice.
Read more about Regulatory Requirements for the Validation of Analytical Methods in your GMP Compliance Adviser, the most comprehensive GMP online knowledge portal worldwide.
Proof of the suitability of analytical test methods is a general GMP requirement. While the EU GMP Guideline Part I generally requires "validated test methods", Part II refers to the ICH guidelines with regard to the content of method validation. 21 CFR Part 211 also requires in §194(a)(2) that the suitability of the test methods must be verified.
The ICH Guideline Q2A on validation of analytical procedures published in 1994 was very important for the standardization of terms and definitions as well as for establishing the basic requirements for analytical method validation. The guideline was adopted by the EU in 1995 as a scientific guideline in the Notice to Applicants (EudraLex Volume 3) and is therefore binding for new authorizations. In 2005, the guidelines ICH Q2A and Q2B were merged under the name Q2(R1) with the new title Validation of analytical procedures: Text and Methodology, without changes to their content.
Since the 1990s, significant developments have occurred, particularly in the area of pharmaceutical production, such as quality-by-design tools, risk analysis and lifecycle. The ICH validation guideline Q2(R1) focuses mainly on chromatographic test methods. Due to the lack of acceptance criteria, the basis to prove the required suitability is diffuse, and the chapter on "Linearity" confuses the response function (calibration model) with the linearity of the analyte in the sample, i.e. with the precision. The latter has led to problems with analytical techniques which display non-linear response functions, especially with biological methods and products.
In November 2018, the ICH therefore published a concept paper on the revision of the validation guideline and the introduction of a new guideline Q14 on analytical procedure development. After a period of almost two years, the drafts of the two guidelines were published for public consultation in March 2022 in step 2 and each received more than 3,000 comments. At the ICH meeting in Prague on 31 October - 1 November 2023, the finalized guidelines in Step 4 were adopted by the regulatory ICH members, but were not published on the ICH website until 20 December 2023 (valid since June 2024).
Some of the shortcomings were rectified, but as expected, the international coordination process also led to various compromises and inconsistencies. The following table summarizes the significant changes and shortcomings from the author's perspective.
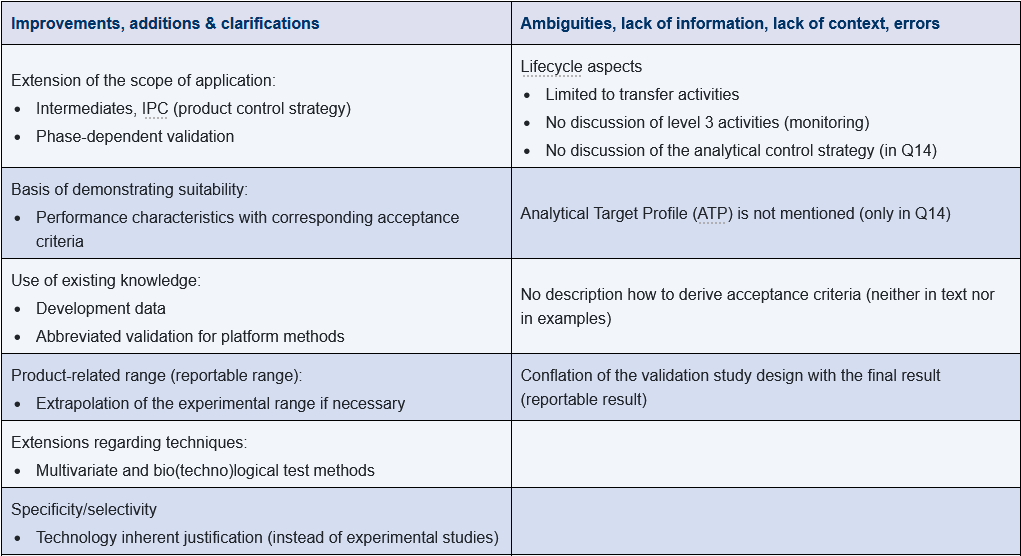
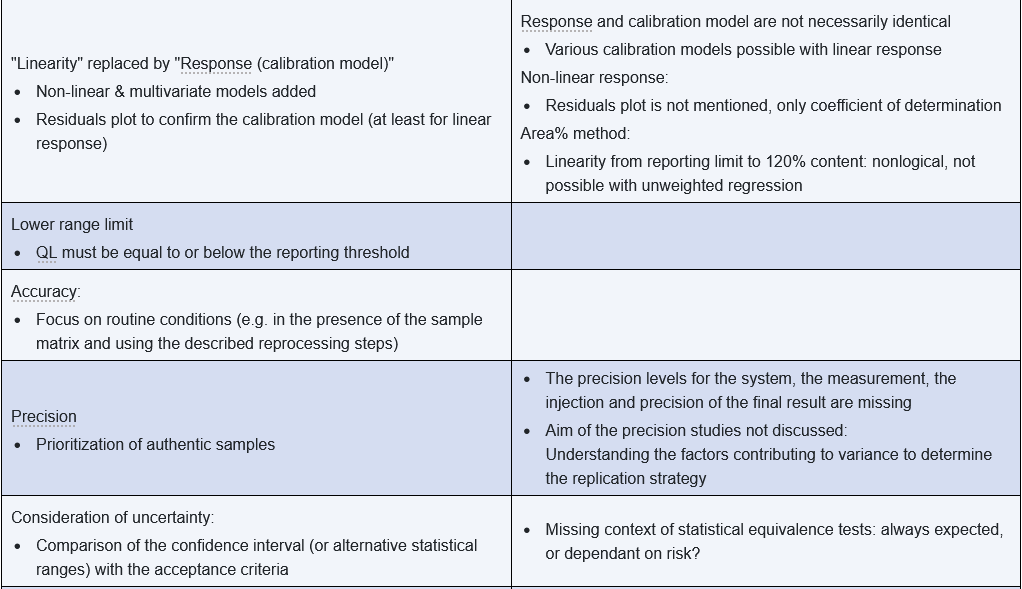
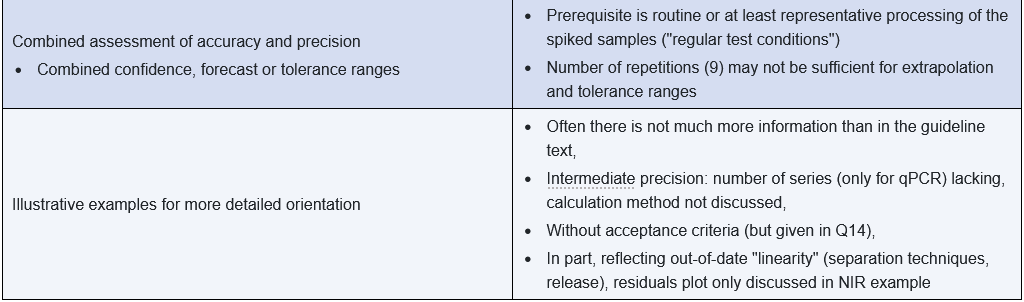
In part 2 of this article, you can read about important aspects of putting the regulatory requirements into practice.
Do you have any questions or suggestions? Please contact us at: redaktion@gmp-verlag.de







 | Named User Licence | 12M")

