
Process Development as the Basis for Process Validation
7 min. reading time | by GMP-Verlag
Published in LOGFILE 35/2021
A GMP compliant and successful process validation is only possible when a 'robust' 1 pharmaceutical development of the medicinal product has been performed, regardless of which development methods are applied.
The meaning of the term 'robust' can be explained best using a process model of the manufacturing process:
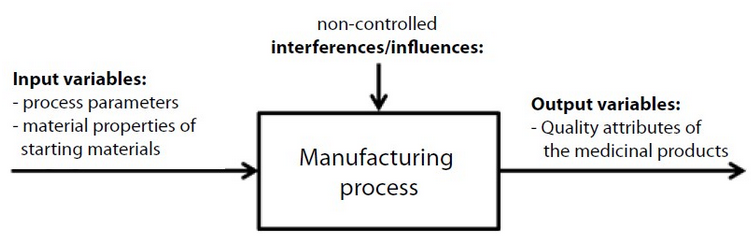
Figure 1 Process model for manufacture of a medicinal product
A manufacturing process can thus be judged to be robust if, in spite of variabilities in the input variables within the permitted ranges (process parameters, characteristics of the raw materials) and the unavoidable interferences, it is possible to yield the medicinal product dependably which exhibits the quality attributes (output variables) which also vary only within the predefined specification limits.
The first step in pharmaceutical development is thus to identify the so-called critical quality attributes (CQA) of the medicinal product based on the quality target product profile (QTPP) or other existing product knowledge. The CQAs form the basis for developing the formulation and process.
Critical quality attributes (CQAs) are all characteristics of a medicinal product or raw materials and intermediate products which are to be maintained within certain limits or distribution ranges 2 to ensure the efficacy and safety including safe usage of the medicinal product. CQAs can reflect physical, chemical, biological and microbiological characteristics. The determination of their criticality is performed using risk assessments. The influence of variability of this characteristic on the patient's health is evaluated (CQAs are not immediately related to the manufacturing process, rather they are primarily clinically defined parameters; CQAs are most frequently laid out in the product specifications at the conclusion of the medicinal product development).
The following greatly simplified graphic may serve to illustrate the relationship:
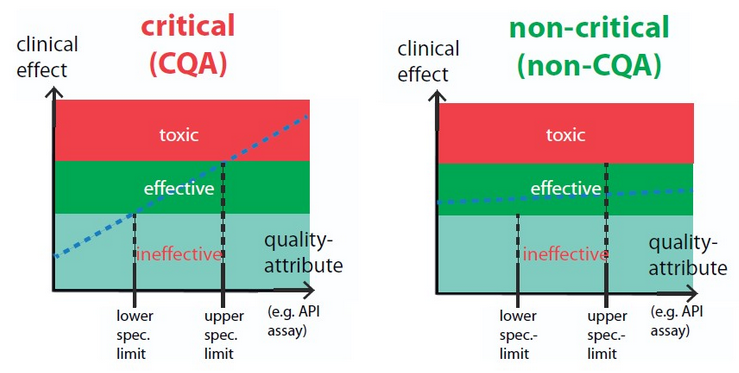
Figure 2 Critical vs non-critical quality attributes
Based on the CQAs 3, the quantitative formulation of the medicinal product and the manufacturing process can then be developed (quality of the raw materials being used, manufacturing technology, equipment design, process steps, process parameter settings, and controls to perform).
With regard to the ability to validate the process, experimental trials play an essential role in the development and optimization of the process to determine
- if the intended parameter settings can be maintained reliably and
- which influence variations of the process parameters and the raw material properties have on the medicinal product’s critical quality attributes.
Critical process parameters (CPPs) and critical material attributes (CMAs) represent in a sense these parameters for which variations can lead to changes in one or more CQAs of the medicinal product, which explains why these CPPs and CMAs must be controlled and monitored.
It follows that 'non-critical' process parameters and material attributes are those for which their variability has no relevant influence on the CQAs of the finished drug product:

Figure 3 Critical vs non-critical process parameters (simplified)
The knowledge gained about the criticality of the quality attributes, process parameters and material attributes should be reflected during the design of the control strategy 4, which is to be prepared for the control and monitoring of the manufacturing processes (specifications of raw materials and intermediates, type and frequency of in-process and material controls, interventions to control the manufacturing process, manufacturing specifications, permitted acceptance criteria for all parameters tested etc.).
All the development tests performed in this framework are typically performed at lab scale, i.e. using batch sizes which are much smaller than the later commercial production sizes planned for manufacturing.
It is possible that the knowledge gained about the influences of process parameters and material properties and their impact on critical quality attributes of the drug product are not transferable to the commercial scale production in the same manner.
The converse is also possible that risks develop at commercial scale which were not found at the lab scale. 5
For this reason, another aspect covered during pharmaceutical development is the so-called scale-up procedure, i.e. investigations of the effect that changes in batch size have on the ability to uphold critical quality attributes. It may be identified that process parameters behave in a scale-dependent or independent manner. Accordingly, the process or material attribute specification limits may need to be adapted as a result of the knowledge gained during scale-up.
For this and for other 6 purposes so-called pilot batches are manufactured and tested (batch size at least 1/10 of the commercial batch size, for solid oral dosage forms at least 100,000 dosage units).
At this development stage process validations are often conducted at the pilot scale.
Potentially it may be necessary to manufacture and test commercial scale trial batches as part of the technology transfer from development to the commercial production site prior to initiation of routine operation and to test also for further effects of scale.
When approaching the point of marketing authorisation and the first placing on the market, the development activities occur under increasingly stringent GMP conditions producing the corresponding documentation on the one hand and because the batches produced are often used in clinical studies, on the other hand, to ensure that the submitted registration documents for the manufacturing quality control and stability can be reproduced during commercial manufacturing.
The result of the development activities is the socalled process design, comprising a defined commercial manufacturing process and a defined control strategy for this process (including, for example the master batch record, in-process control standards, specifications and the appropriate test methods).
All the knowledge gained during development, optimisation, scale-up and transfer of the manufacturing process should be reflected in the design.
The knowledge included in the process design should be made available to the production site to enable a well-founded validation of the manufacturing process.
Checking the appropriateness of the original process design and the control strategy is the responsibility of the competent authority. Since the process design and control strategy may be changed over the product lifecycle, for which notification of or approval by the competent authorities are not always mandatory the appropriateness of changes made may also be subject to GMP investigation.
1 Annex 15 new, Section 5.1
2 Distributions: e.g. particle size of powders for inhalation or oral dosage
3 In this figure a simplifying assumption is made that the CQAs are known and unchanging from the beginning of the medicinal product´s development. In practice, over the course of development increasing knowledge (esp. as a result of pre-clinical, clinical and pharmaceutical-analytical study results) makes it necessary to update the CQAs and thus make changes to the product and process. Especially the development of new medicinal products does not follow a direct path, as shown here.
4 In this figure a simplifying assumption is made that the CQAs are known and unchanging from the beginning of the medicinal product´s development. In practice, over the course of development increasing knowledge (esp. as a result of pre-clinical, clinical and pharmaceutical-analytical study results) makes it necessary to update the CQAs and thus make changes to the product and process. Especially the development of new medicinal products does not follow a direct path, as shown here.
5 For example, during tablet compression the tablet blend may segregate in the container as a result of vibrations during the long running time on the tablet press.
6 other purposes: provision of samples for stability testing, pivotal or bioavailability/ bioequivalence testing and pivotal clinical studies
Do you have any questions or suggestions? Please contact us at: redaktion@gmp-verlag.de






 | Named User Licence | 12M")

