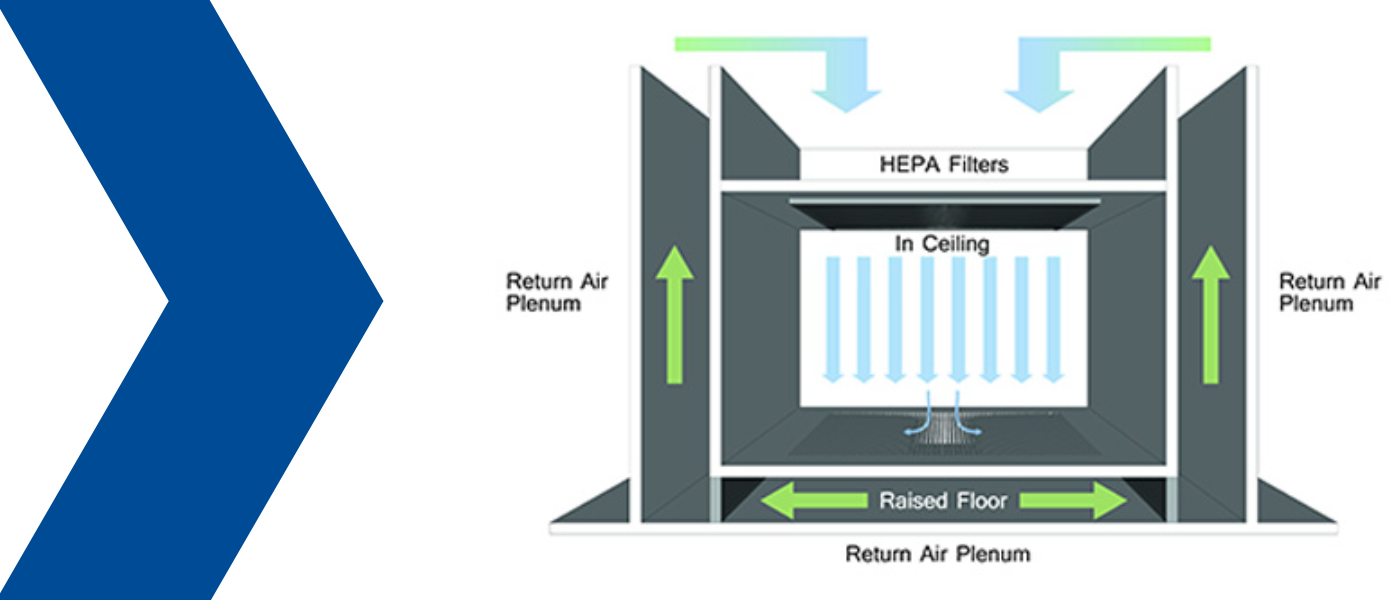
Test interval for integrity testing of HEPA filters in a laminar flow above an injection moulding machine
12 min. reading time | by Harald Flechl
Published in LOGFILE 34/2021
Question:
We operate a laminar flow above an injection moulding machine in a cleanroom, GMP Grade C (ISO 8 in operation, ISO 7/at rest). The laminar flow achieves ISO class 5 within the cleanroom area.
How often should the terminal HEPA filter integrity test be performed? Is there a default? Or do we need to perform a risk analysis and determine the intervals based on this?
Response:
Recommendation summary
A) If the (commercial) operating license was granted according to purely legal requirements, a documented risk assessment is recommended in any case to justify the inspection intervals.
B) If, in addition to the legal requirements, the operating license is also in accordance with EU GMP guidelines, it is sufficient, until the revision of Annex 1, to carry out a documented risk assessment to justify the test intervals (see under "What points should the risk assessment cover? ").
On the part of the authorities, however, a semi-annual inspection is expected for ISO class 5 (laminar flow is equated with GMP Grade A). In the revision of Annex 1 (draft 2020), these intervals are already described (see under "Which GMP regulations and standards apply here?").
Recommended test interval for HEPA filter integrity testing
- During initial installation or replacement of the filter
- Recurring inspection for new filters after one year, thereafter after 3 years, if no other event as per EN ISO 14644-3, Annex A.3, a) to d) occurs beforehand (see section "Which GMP regulations and standards apply here?")
- When the life expectancy specified by the manufacturer is reached under the given operating conditions, the interval should be reduced to annually or semi-annually (result of the risk assessment).
- When particle contamination due to airborne contamination is detected on the product.
What points should the risk assessment cover?
A reference to the periodic review could be included in the operating permit. The legal requirements according to which the permit was issued should also be apparent there.
If the facilities and the production operations are approved and conducted in accordance with GMP, the legal requirements still provide the primary governance. Although the GMP guidelines are legally considered a "recommendation", the recommendations of the guidelines are considered a "requirement" by the authorities/inspectors and are also evaluated as such. Since an injection moulding machine is involved, the Medical Devices Directive may also be authoritative, depending on the product manufactured and its use (e.g. as an "accessory to a medical device"). In addition, the EU GMP Guide, whereby "aseptic" production can be excluded, can form the basis of the approval, whereby "non-sterile production" also does not specify Grade A (ISO 5) or B. The GMP guidelines in turn refer to the ISO 14644 series of standards for cleanroom-related topics.
This means: If the approval was granted under the EU GMP Guide, one should use the "requirements“ of the GMP Guide in addition to the ISO 14644 series for the assessment. If this is not the case, i.e. a purely "commercial" operating permit is in place, one can only use the ISO guidelines as a basis for assessment.
All guidelines have one thing in common when setting an inspection interval: the risk should be assessed.
In a risk assessment for the evaluation of a leak in a HEPA filter (class H13 or H14 according to EN 1822), the following points would have to be considered (no claim to completeness):
- Most important: What effect does a potential leak in the filter have on my product? I.e. what is the risk for the person using this product in case of potential contamination due to a leak in the HEPA filter? (In the extreme case, the result would be: No cleanliness class ISO 5 is required - this makes the other points obsolete!)
- What is the contamination of the air immediately upstream of the HEPA filter during normal operation? Are pre-filters used, if so, what is the separation efficiency of these filters? (Used to assess what effect a leak in the HEPA filter can have. Studies have shown that in the event of a leak, approximately 3% of the particles upstream of the HEPA filter can penetrate through the leak.)
- A leak in a HEPA filter does not occur by itself, but mostly due to mechanical stress/damage when handling the filter during installation (detected during acceptance measurement = function test). Is there a risk of filter damage when manipulating in the tool area (tool exchange, cleaning, etc.)? If this can be the case, a leakage test would be advisable according to the risk assessment.
- After a certain operating time of the filter and depending on the ambient parameters (temperature, humidity), the sealing compound with which the filter package is sealed into the frame can become "brittle" and air-permeable. At "normal" temperatures (approx. 19°C to 25°C, approx. 35% to 65% relative humidity), a filter service life of 5-10 years can be expected - the filter supplier can confirm this more precisely (shelf life). Only after this time could a leak possibly be detected (Note: I know of HEPA filters in Grade A and B that have been in operation for 20 years.)
- The leak measurement (integrity test) of a HEPA filter is not a science, but the measurement technician needs appropriate experience and the right measurement equipment. Approx. 80% of detected leaks are due to incorrect measurements (induction; incorrect measuring arrangement; probe diameter does not isokinetically match the actual airflow; leak tests are carried out as an "efficiency measurement"; or similar).
- Within the "laminar flow" area one should know the airflow visualization by "flow visualization" to determine in which area a leak could be critical for the product or if thermal buoyancy leads to a backflow in the critical area.
- Thermal buoyancy can also be the cause of detected contamination and then has nothing to do with the efficiency of the filter (leakage) (cf. point 6).
- Filter contamination (dust entrapment) in the filter is due to the purpose of the filter and is not a "deterioration in quality". On the contrary, a higher dust load of the filter increases the efficiency of the filter somewhat. However, the contamination increases the pressure drop caused by airflow and is then an energetic problem (the increase up to the bursting pressure of the filter is usually not achieved by the fans).
- In the case of a "laminar flow" system above an injection moulding tool with a high temperature, the widely assumed "standard velocity" of 0.45m/s (± 20%) is usually insufficient due to the thermal lift that occurs and is often set at 0.60m/s or higher. In this case, the filters are also not operated with the "nominal air volume" at an incident air velocity over the filter nominal surface [e.g. 610 x 610mm = 0.37m²] of 0.45m/s but are usually operated with a higher air volume. If the airflow rate is higher than the nominal airflow rate specified by the filter manufacturer, the filter effectiveness (separation efficiency) is reduced. A filter of class H14 could then only correspond to a filter class H13, but this will hardly have any effect on the air cleanliness. Important here: The particle probe for leakage measurement must be adapted to this air velocity (see point 5).
Which GMP regulations and standards apply here?
In the currently valid Annex 1 (2008) of the EU GMP Guideline, the monitoring of clean rooms and clean air systems is described as follows:
8. Clean rooms and clean air devices should be routinely monitored in operation and the monitoring locations based on a formal risk analysis study and the results obtained during the classification of rooms and/or clean air devices.
9. For Grade A zones, particle monitoring should be undertaken for the full duration of critical processing, including equipment assembly, except where justified by contaminants in the process that would damage the particle counter or present a hazard, e.g. live organisms and radiological hazards. In such cases, monitoring during routine equipment setup operations should be undertaken prior to exposure to the risk. Monitoring during simulated operations should also be performed.
The Grade A zone should be monitored at such a frequency and with a suitable sample size that all interventions, transient events and any system deterioration would be captured, and alarms triggered if alert limits are exceeded. It is accepted that it may not always be possible to demonstrate low levels of ≥5.0 µm particles at the point of fill when the filling is in progress, due to the generation of particles or droplets from the product itself.
The draft Annex 1 (2020) - which is not expected to be published until the end of 2021 and will come into force in 2022 - proscribes a periodic review:
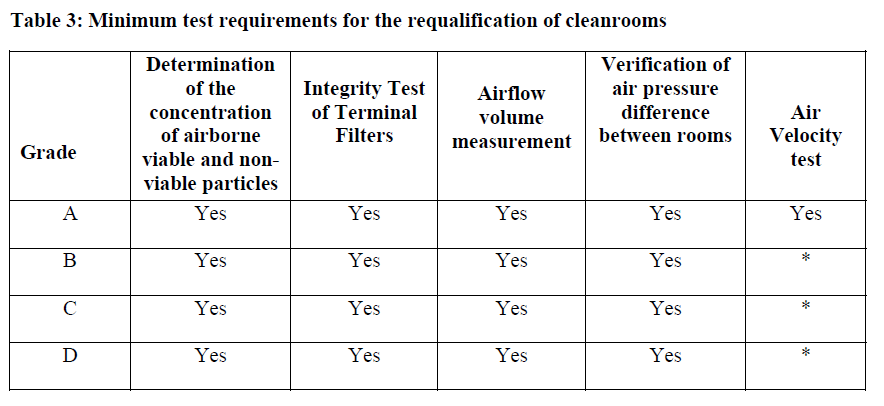
Intervals for the test:
For Grade A (ISO 5) & B, the maximum interval for "requalification" is 6 months and for Grade C & D, the maximum interval is 12 months.
EN ISO 14644-2:2016 describes in clause 5:
5. Periodic classification of air cleanliness by particle concentration
Periodic classification testing shall be undertaken annually in accordance with ISO 14644-1. This frequency can be extended based on risk assessment, the extent of the monitoring system, and data that are consistently in compliance with acceptance limits or levels defined in the monitoring plan.
NOTE: ISO 14644-3 specifies ancillary tests related to other aspects of cleanroom performance such as pressure difference, airflow, etc.
EN ISO 14644-3:2020 describes in the annex:
A.3 Scheduling of tests and retests. At a minimum, tests should be conducted as follows:
(a) according to the classification following ISO 14644-1;
(b) during the retest during the initial commissioning;
(c) during the retest after detection and correction of faults;
(d) during the retest after a modification;
(e) during the periodic retest.
Risk assessments should be carried out to determine appropriate intervals for periodic tests. Monitoring data, trends and test results should be used to confirm and, if necessary, adjust the time intervals for the selected tests. (Quote from the EN ISO 14644-3:2020 is an internal translation from the German standard.)
Do you have any questions or suggestions? Please contact us at: redaktion@gmp-verlag.de






 | Named User Licence | 12M")

