
Insufficient traceability of the batch documentation
Excerpt from the GMP Compliance Adviser, Chapter 21.C.2.7
12 min. reading time | by Dr. Sabine Paris
Published in LOGFILE 11/2021
The deficiency
Time specifications for the start of production (preparation from 8:30) and for the end of production (15:30) were entered in the batch documentation.
However, microbiological monitoring in cleanroom Grade A during sterile production was only carried out between 11:00 and 15:00. It was not possible to determine from the batch documentation whether this covered the critical work steps.
In addition, the batch documentation could not sufficiently reconstruct the packaging material reconciliation, as the incoming package inserts were given in kg and the outgoing package inserts as number of pieces.
(Ref.: EU GMP Guide Part I No. 4.8 and 4.20)
That was the problem
For sterile production in cleanroom Grade A, microbiological monitoring is required during production. The monitoring should be carried out on a risk-based basis, especially during critical process steps (e.g. product is exposed to the ambient air for a "long" time). Annex 1 of the EU GMP Guide contains concrete requirements for this (see C.4.1 Annex 1 Manufacture of Sterile Medicinal Products).
The company had carried out a risk assessment and classified the process steps into "subject to monitoring" and "not subject to monitoring". However, the microbiological monitoring by means of sedimentation plate and active airborne microbial collection was only carried out over part of the complete process time. It was not apparent from the batch documentation that the critical process steps assessed as requiring monitoring had been carried out exclusively within this monitored time window between 11:00 and 15:00. Only the start and end of production were specified in the batch documentation, but these were both outside the monitored time window. This meant that it was not possible to verify whether all critical work steps were covered by microbiological monitoring. For the critical steps of aseptic production it could not be documented that the microbiological limit values had been complied with.
This is the most common pitfall
Batch documentation is a document that is very often in the hands of the employees carrying out the work and those with "inspection or review functions". The more often a process is performed, the more experience the employees have, but the more it becomes "routine". In everyday operation, not every specification or requirement can and will be questioned.
As a result, the traceability of the batch documentation is lost at one point or another. This is often particularly noticeable with regard to the points shown in Figure 1 and Figure 2:
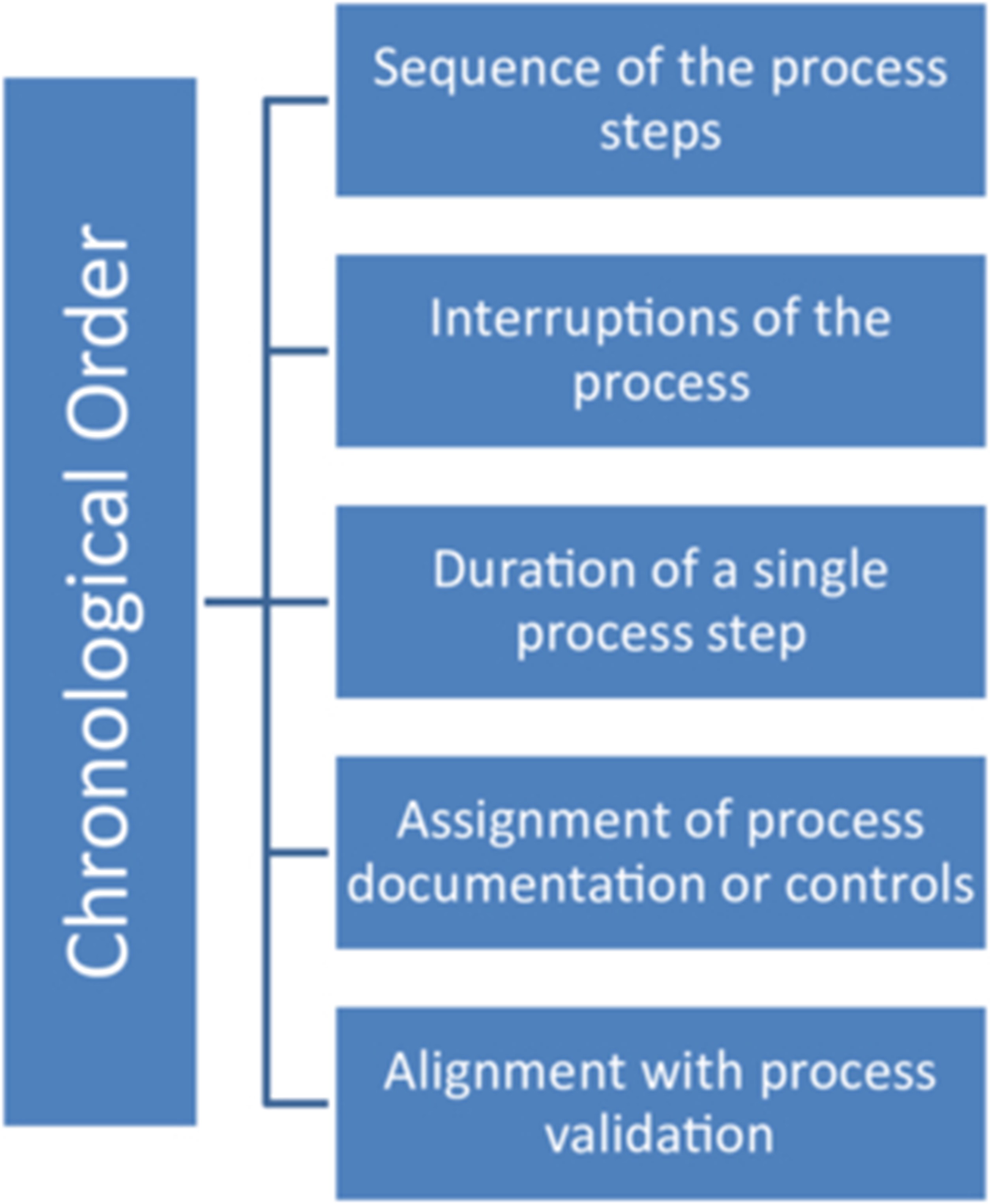
Figure 1 Details of the chronological order, which are often not sufficiently traceable in the batch documentation
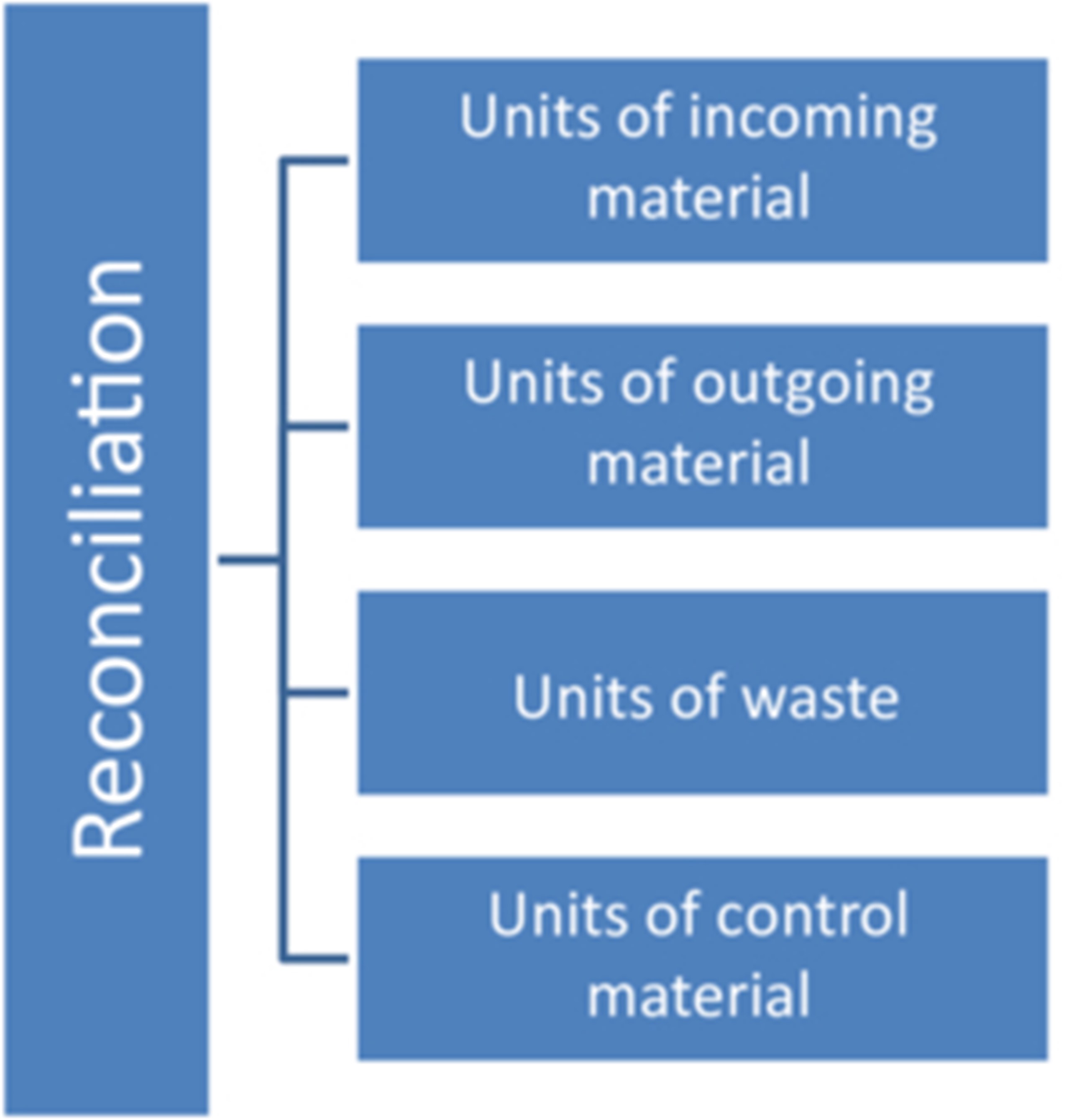
Figure 2 Reconciliation problems that complicate traceability
To avoid this error
The manufacturing instruction and the batch documentation are prepared at the latest at the start of process validation. After process validation, they are usually adjusted finally. Preparation and adaptation are also usually carried out with great care. Nevertheless, discrepancies or errors often remain.
Such discrepancies or errors in the batch documentation can usually only be detected by "independent" third parties, since the employees who work with it on a daily basis routinely "unwind" the "usual program".
In the audit, these persons should pay particular attention to the full traceability of individual steps. Traceability is particularly important
- for points whose (chronological) order is relevant, and
- between the batch documentation and supplementary printouts or documents of the batch documentation (instrument printouts, cleaning labels, particle monitoring, microbiological monitoring, temperature, humidity, differential pressures, etc.).
It is better to ask too much once, than too little once. Even if the production staff may not like it. It might have been noticed in the above described deficiency that although microbiological monitoring is carried out over a documented time window, a comparison with the time window of critical activities in the batch documentation is not possible.
|
|
 Batch documentation must be traceable!
Batch documentation must be traceable!
This means that sometimes uncomfortable questions may be asked, e.g. by internal auditors – and the necessary (personnel) resources must be provided.
If individual steps are not traceable, e.g. with regard to the chronological order, it must be assessed whether they are GMP-relevant:
- For example, is the duration of the encapsulation step GMP-relevant, because the capsules are thus exposed to the humid ambient air for a longer period of time and can therefore more or less stick together?
- Can the information on the duration of the process interruption be waived, even if only a process interruption of 24 h was validated in the process validation?
Attention should also be paid to the consistency of different product-related documents with each other, especially process validation, validation of holding times, media fills or clean and dirty hold times with the information in the batch documentation.
The reconciliation in the course of the batch documentation is also often problematic. This is often due to the fact that different materials that are included in the reconciliation sheet are calculated in different units for incoming and outgoing materials or "pro forma data" is taken from delivery documents.
For example, incoming patient information leaflets often contain either a weight specification or the pro forma specification of the delivery note with e.g. 1000 leaflets. In fact, 1005 or even only 991 leaflets may have been delivered. On the output side, however, the used instructions for use are usually counted individually, so that an exact comparison is difficult. Here, an attempt should be made to align the units in a meaningful way. Information that is taken from delivery notes, for example, should be marked accordingly. This is not "correct" information that can be used for precise reconciliation.
If an "accurate" reconciliation cannot be drawn up, the associated risk should be assessed and measures taken if necessary.
| To ensure the traceability of the batch documentation |
|
Figure 3 Important measures for the traceability of batch documentation
Do you have any questions or suggestions? Please contact us at: redaktion@gmp-verlag.de




 – Not Just an Extended Site Master File")


 | Named User License | 12M")

