
Filter integrity test
Excerpt from Chapter 12.E.4.3 Filter integrity test of the GMP Compliance Adviser and GMP Series Aseptic Processing
7 min. reading time | by Ruven Brandes
Published in LOGFILE 30/2020
The integrity of the filter must be checked before and after use. This is a requirement of the EU GMP Guide1 and of the Ph. Eur. 2 The FDA3 also requires the integrity check, focussing on performance after the filter has been used.
The check for integrity before use remains optional as specified by the FDA. The filters should be installed before being tested. With the test equipment available on the market, this is possible without further ado.
There are several tests that can be used to check the properties and correct functioning of membrane filters:
- Bubble point test
- Diffusive flow/forward flow test
- Pressure hold/decay test
Bubble point test (B.P. test)
In the B.P. test the pressure is determined at which the transition of the diffusive gas flow to the free flow via pores in the filter material which are no longer wetted takes place. With this process, the gas pressure is increased continuously or in stages on the unsterile side, and a check is made to see when the pressure decreases disproportionately.
Diffusive flow test
In the diffusive flow test, the measurement is always made applying a pressure of ca. 70 to 80% of the bubble point pressure. Various measurement methods can be used for the subsequent evaluations.
Pressure hold test
When performing the pressure hold test, pressure is applied to the unsterile side of the filter, which is then closed. The gas can then only escape through the wetted membrane as a result of diffusion processes. This gas flow leads to a drop in pressure, which can be measured and compared with a defined maximum limit value. Here it is above all important to know the precise gas volume on the unsterile side.
In the diffusive flow test, a boundary diffusion is measured using the drop in pressure. To permit a boundary diffusion to be assessed with the help of the drop in pressure, the net volume on the input side (volume on the unsterile side) must be known precisely. In addition to the drop in pressure, the volume is therefore also determined. To do this, the test pressure of the filter system is released into a known reference volume. The resultant drop in pressure permits the unknown volume to be calculated in accordance with Boyle-Mariotte's law. This volume and the drop in pressure are then used to calculate the diffusive flow, and this is compared with the system-independent diffusive threshold of the filter element which is to be tested.
All tests are to a large degree dependent on the surface tension of the wetting liquid. When liquids with different surface tensions are used for wetting, e.g. water, alcohol, disinfectants, etc., different values are obtained as a result. Consequently a decision should first be taken regarding whether a product-specific integrity test should be conducted or an integrity test using a standard medium (e.g. water). In the case of a product-specific integrity test, the filter manufacturers are the direct contacts. They develop a product-specific bubble point test.
Loss of filter integrity
When a filter integrity test fails, the product should, if possible, be filtered again. If this is not possible and the filter which failed the test was the "police filter", this can only result in the batch being discarded. This has to be done even if the test for sterility was passed successfully, because the test for sterility is only a "snapshot" and is statistically not meaningful.
The pressure range recommended by the manufacturer is used when testing filters. In an intact system the pressure will remain constant over a specific time period. Depending on its appearance on the pressure curve printed out, the drop in pressure can have various causes.
Filter damage can be caused by:
- Pressure bumps
- Unsuitable sterilisation conditions (e.g. condensate formation in the filter candle or too high a pressure differential)
- Mechanical damage (e.g. during assembly)
The failure of a filter integrity test needs not always be due to a damaged filter. A test can also fail for the following reasons:
- Filters not properly rinsed
- Filters not wetted completely
To exclude such faults before a filter is classified as damaged, in its Technical Report No. 26 the PDA has established an Integrity Test Troubleshooting Guide (see Figure 12.E-3 ). This provides helpful instructions on how to prevent the aforementioned faults. The filter manufacturers frequently provide their own Troubleshooting Guide in their instructions, which is tailored to the properties of the specific filter.
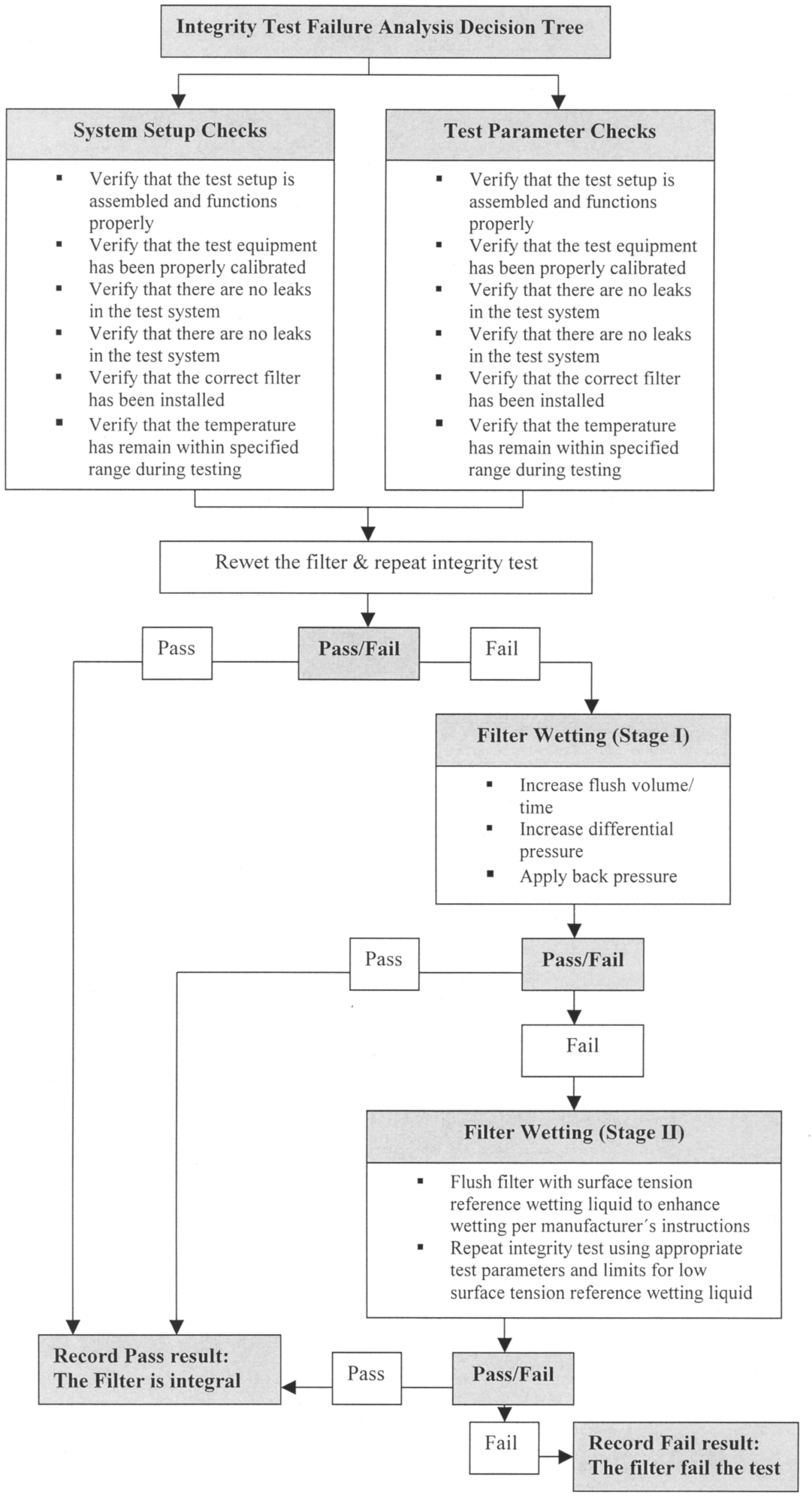
Figure 12.E-3 Integrity Decision Tree (from: PDA Technical Report No 26)
Sterilisation of filters
Regardless of whether a sterile filter is reused (see 12.E.4.2 Materials, designs and use of filters) or not, the sterile filter must be sterilised before use.
When steam sterilisation is employed, the manufacturer's instructions must always be observed. Sterilisation is normally performed at 121 °C, with sterilisation times of 20 to 30 min. being regarded as appropriate. Some candles can be sterilised at 134 °C or 145 °C for 30 min.
When the filters are sterilised, they must be completely flushed with steam. If this is not fully achieved, air cushions are created. Overheated steam, which does not achieve total sterilisation efficiency, forms at these places. The steam quality must also be checked in order to exclude the influence of non-condensable gases.
The following solution approaches are possible:
When sterilisation is performed in an autoclave, validated pre-vacuum cycles are indispensable.
In the case of SIP, valves must be arranged in such a manner that removal of the air cushions is reliably guaranteed by flooding.
The placement of bio-indicators in validation runs is an essential recommendation.
The effectiveness of the filter sterilisation process in situ is frequently influenced by condensate formation in the cartridge. The condensate blocks the pores and may hinder the steam flow through the wetted surface. In this case the filter is heated unevenly. This can be solved by heating the filter slowly and checking that the steam pressure is maintained at the outlet.
- EU GMP Guide, Annex 1, Section 113
- Ph. Eur. Edition 6, Basic Volume 2008, 5.1.1 Methods for manufacturing sterile preparations, p. 769
- FDA Guidance for Industry, Sterile Products Produced by Aseptic Processing-Current Good Manufacturing Practice, Part V Personal Training, Qualification, Monitoring, Part B. Filtration Efficacy; 2004 (see D.10 Guidance for Industry Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice)
Do you have any questions or suggestions? Please contact us at: redaktion@gmp-verlag.de






 | Named User Licence | 12M")

