
MHRA: Feedback from GMP inspections
15 min. reading time | by Tim Sandle
Published in LOGFILE 07/2020
To assist pharmaceutical manufacturers and distributors to understand the areas where good manufacturing practice (GMP) inspectors have found compliance problems during GMP inspections in the UK and overseas, the UK Medicines and Healthcare Products Regulatory Agency (MHRA) GMP Inspectorate has issued data, during October 2019, relating to common deficiencies from previous GMP inspections conducted during 2018 [1].
While the anonymised raw data provided by the GMP Inspectorate is of general interest, additional analysis is required to draw meaningful inferences. In this article, the data has been reviewed and presented, in order to obtain an overview of key trends.
Based on a review of critical and major deficiencies, pharmaceutical manufacturers need be most concerned about the following.
-
Quality risk management.
-
Contamination control strategy, both microbial and cleaning validation.
-
Environmental monitoring.
-
Overall attention to the quality system, especially in relation to change controls and deviations.
-
Appropriate and thorough investigations.
These represent the common trends for the year 2018 among over 6000 citations across almost 300 inspections, and provide focal points for driving internal improvements. In general, sterile manufacturers receive a far greater number of deficiencies compared with non-sterile manufacturers.
Classification of deficiencies
The MHRA conducts product-related GMP inspections when assessing an application for a UK marketing authorisation. Site visits invariably include each part of the facility or process involved in producing, purchasing and distributing medicines, including the following.
- Manufacturing areas.
- Quality Control (QC) and microbiology laboratories.
- Stock and stock management.
- Storage areas.
- Temperature monitoring.
- Returns areas.
- Purchasing and sales functions.
- Transportation arrangements.
During the inspection, observations are made, many of which are then assessed as deficiencies according to European Medicines Agency (EMA) requirements. Deficiencies are classified as follows [2].
Critical deficiency
- A deficiency which has produced or significantly risks producing a product which is harmful to humans or veterinary patients or which could result in a harmful residue in a food-producing animal.
- Any departure from good distribution practice (GDP) that results in a significant risk to patients. This includes an activity which increases the risk of counterfeit medicines reaching patients.
Major deficiency (a non-critical deficiency)
This is a deficiency which includes at least one of the following.
- Has or may produce a product that does not comply with its marketing authorisation.
- Indicates a major deviation from GMP or GDP or from the terms of the manufacturer licence or wholesale licence.
- Indicates a failure to carry out satisfactory batch release procedures or (within the European Union (EU)) a failure of the qualified person or responsible person to fulfil their legal duties.
- A combination of several ‘other’ deficiencies which on their own may not be major but together may represent a major deficiency and should be explained and reported as such.
Other
A deficiency which cannot be classified as either critical or major.
Review of 2018 data
From the information provided by the MHRA, there were 6210 observations from inspections of distributorsand manufacturers, classed as either ‘critical’, ‘major’ or ‘other’. The 6000 plus observations related to a total of 286 inspections. The majority of the inspections related to manufacturers, at 93% (see Figure 1).
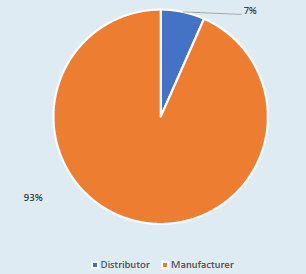
Figure 1. MHRA observations between manufacturers and distributors.
Geographical differences
During the time period, the MHRA inspected pharmaceutical facilities in Bangladesh, China, India, Japan, South Korea, Singapore, UK and USA. The majority of the inspections were, unsurprisingly, conducted in the UK (see Table 1).
The list of countries can be interpreted in different ways. The list could reflect the main manufacturing or distribution areas for medicinal products coming into (or from within) the UK; or the list could reflect the relative risk of different manufacturers in different countries (hence India could be seen as presenting a relatively high risk); or the overseas list may simply reflect companies in one country being grouped together for convenience so that inspectors need only take fewer trips. Perhaps the relative risk aspect is the most likely, given the average number of findings per inspection (see Figure 2).
Korea has the highest mean number of findings per country. This is somewhat distorted by only one company having been audited. Perhaps a more meaningful statistic can be drawn by a comparison where a sizeable number of inspections have beenundertaken, as between India (average of 33 findings per company) compared with the UK (average of 19 findings). This may reflect the relative maturity of pharmaceutical processing in both countries.
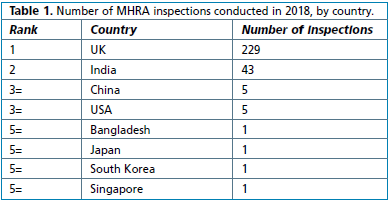
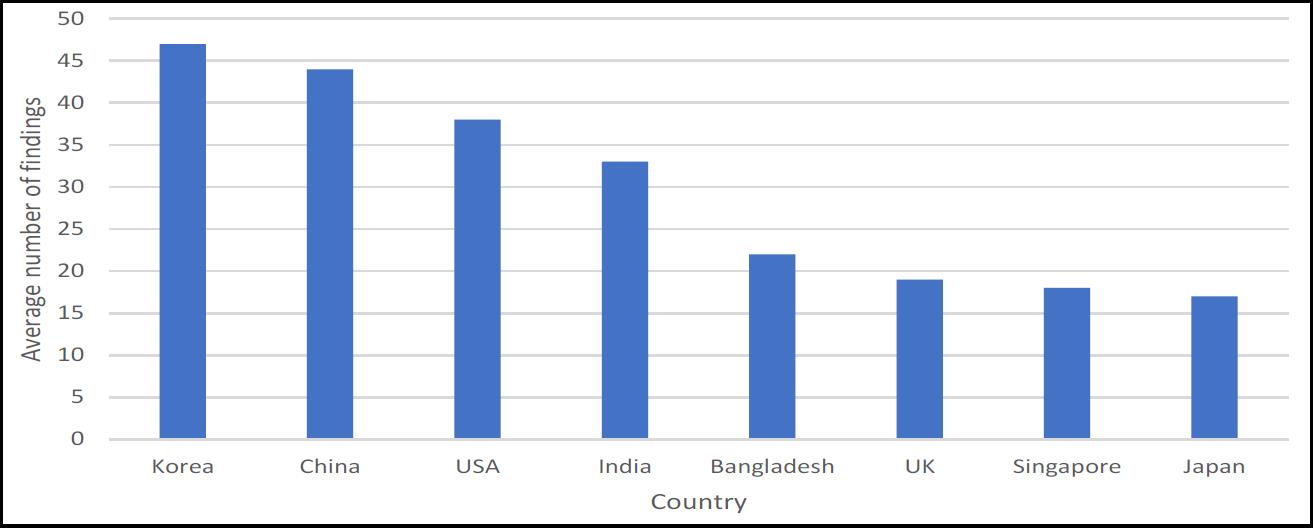
Figure 2. Mean number of MHRA findings per country.
Categorisation of findings
In terms of the categorisation of the deficiencies, the majority were ‘others’ (a category that is often, incorrectly, considered to be ‘minor’ – however, all that ‘other’ infers is that there was insufficient evidence found by the inspectorate to turn the observation into a ‘major’. Hence ‘other’ serves as a warning to the company to look deep into its internal procedures and processes. The breakdown according to inspection category is shown in Figure 3.
From the critical observations, it can be seen that these related to companies in India and the UK only. In India there were 33 critical observations relating to three companies; and for the UK there were 109 observations relating to 11 companies.
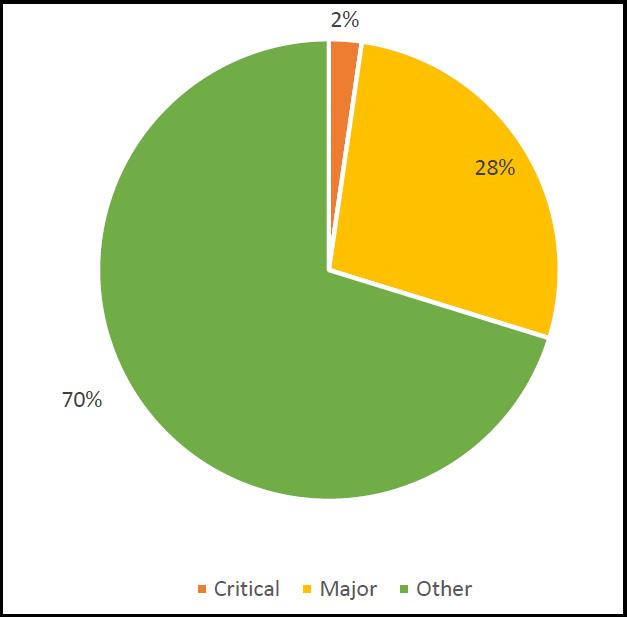
Figure 3: MHRA deficiencies by category – critical, major and other.
Sterile and non-sterile manufacturing
Pharmaceutical products can be divided into sterile and non-sterile. Generally, sterile products are at greater risk in terms of microbiological contamination (due to the route of administration and often reflecting the general health of the patient, such as being immuno-compromised). The majority of GMP observations relate to sterile products. This may reflect the less controlled practices in relation to sterile products manufacturing or it may reflect a greater number of inspections of sterile products facilities (due to the critical nature of the product). The division between these two types of pharmaceutical products is shown in Figure 4.
The relative risks surrounding sterile manufacturing are such that of the 286 inspections, 225 were of nonsterile facilities. These non-sterile facilities accounted for 33% of the observations – an average of nine deficiencies per facility. In contrast, of the 286 inspections, 61 were of sterile manufacturers. These manufacturers accounted for 67% of the observations, or 67 deficiencies per manufacturer. The risk profile is therefore heavily skewed towards sterile production.
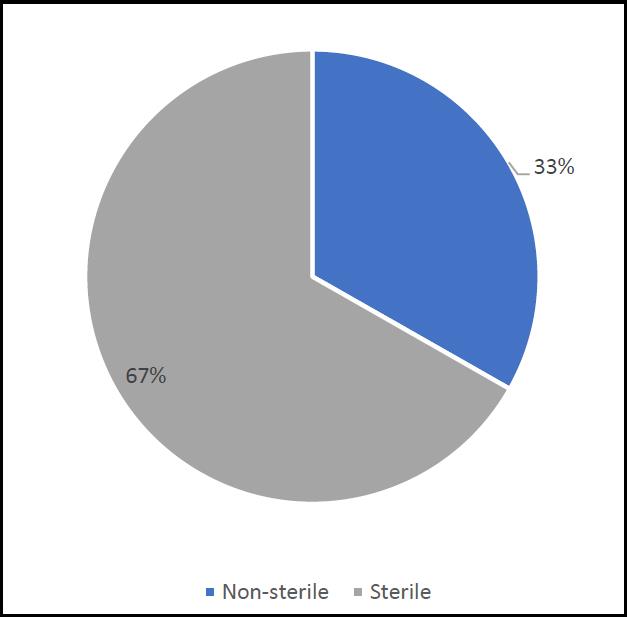
Figure 4. Number of MHRA observations, between sterile and non-sterile manufacturers.
Breakdown by EU Guidelines to GMP Chapter or Annex
The data provides a breakdown of the number of deficiencies according to the chapters that comprise the EU Guidelines to GMP together with the Annexes3. Table 2 provides a breakdown of the chapters, and it can be seen that the most common deficiencies relate to the pharmaceutical quality system; this is followed by documentation (a category that can additionally include some issues relating to data integrity); followed by production and premises, and then quality control. The pie-chart in Figure 5 gives an idea of theproportion of observations relating to each chapter. The pie-chart more clearly shows that around onethird of observations are linked to the quality system, and when combined with documentation, these two chapters account for close to half of all inspection findings.
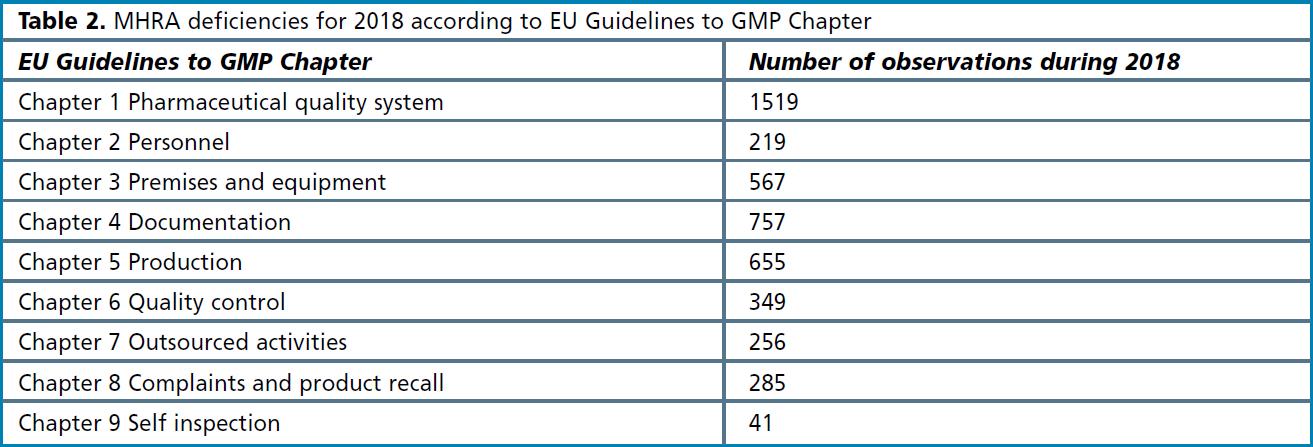
Figure 5. MHRA deficiencies by EU Guidelines to GMP Chapter.
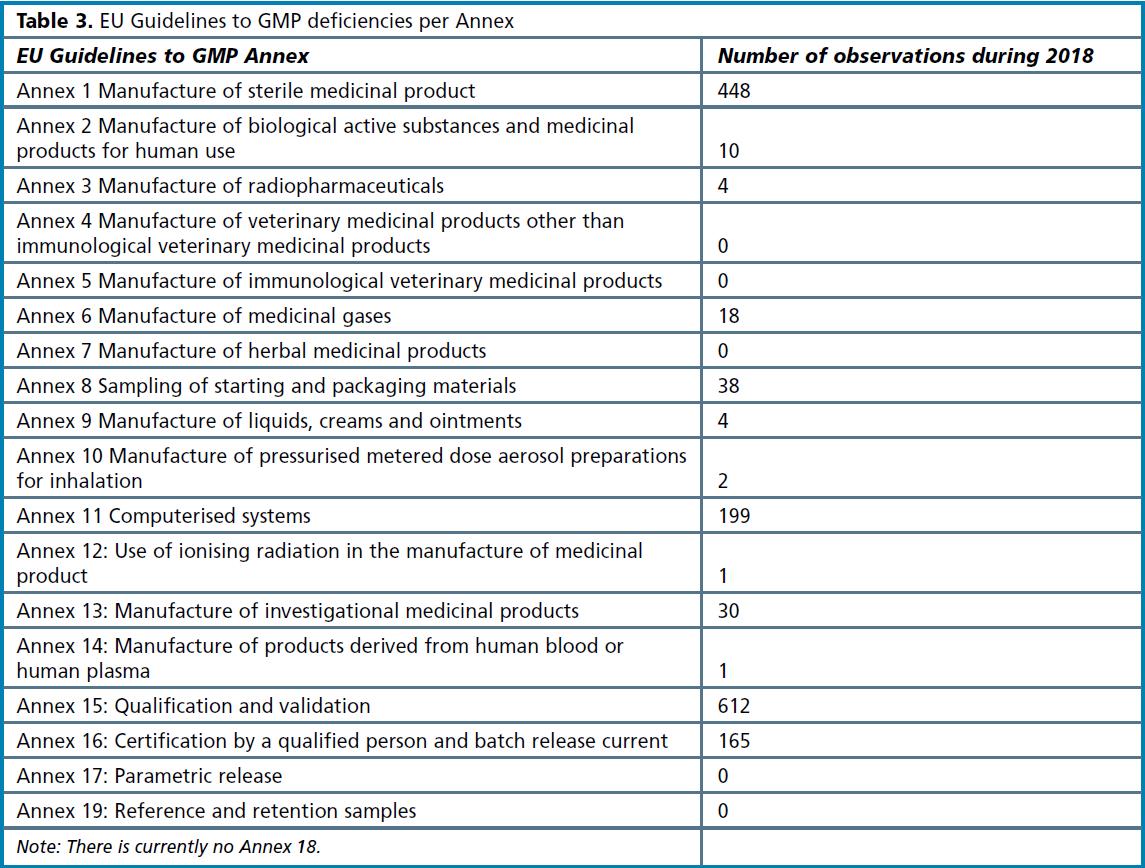
A similar analysis can be made in relation to the Annexes to EU Guidelines to GMP (see Table 3 overleaf). Annex 15 Qualification and validation comes out as the biggest category, followed by Annex 1. While qualification and validation issues relate to all types of pharmaceutical products, Annex 1 has implications for sterile products manufacturers only. This means across all types of manufacturers, 1 in 10 deficiencies were for qualification and validation issues; for sterile products manufacturers, 12% of observations related to Annex 1 deficiencies, making this part of the EU Guidelines to GMP the most common area of concern.
Examination of critical and major findings
The 142 critical findings according to the EU Guidelines to GMP are shown in Figure 6. As the chart indicates, 38% of the critical observations related to EU Guidelines to GMP Chapter 1. This is the same across the categories of ‘major’ and ‘other’, and the data reviewed earlier already points to concerns with the pharmaceutical quality system. The second mostcommon category is Annex 1. This is a more interesting figure, since it indicates that Annex 1 deficiencies are more likely to lead to a critical observation and this again emphasises the concerns around sterile products manufacturing in terms of relative risk. Other EU Guidelines to GMP chapters and Annexes score relatively lower.
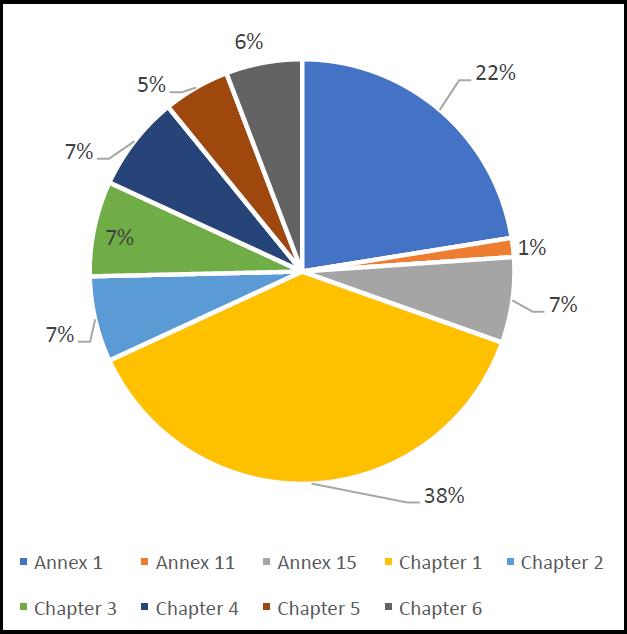
Figure 6. Breakdown of EU GMP critical deficiencies according to EU Guidelines to GMP.
Drilling down the data further (and by crossreferencing to the relevant parts of the EU Guidelines to GMP3), with Annex 1 deficiencies the clauses that were cited most frequently are as follows.
- A1.18 – “Environmental monitoring techniques”.
- A1.20 – “Investigating environmental monitoring excursions”.
Hence, the two most common Annex 1 deficiencies leading to critical observations relate to microbiological practices. Regarding Chapter 1 of the EU Guidelines to GMP, the following were the top two (by some margin).
- C1.4(viii) – “A state of control is established and maintained by developing and using effective monitoring and control systems for process performance and product quality.”
- C1.4(xiv) – “An appropriate level of root cause analysis should be applied during the investigation of deviations, suspected product defects and other problems. This can be determined using Quality Risk Management principles.”
The two most common areas relate to process monitoring and with conducting adequate investigations into deviations, especially relating to root cause analysis. With this latter point, there is a connection to one of the Annex 1 ‘top two’ – investigating microbiological excursions.
For the major findings, the breakdown in relation to the applicable part of the EU Guidelines to GMP is shown in Figure 7. The chart indicates, consistent with the above findings, that Chapter 1 accounts for the greatest number of reported deficiencies. Following this comes Annexes 1 and 15, and Chapter 5.
The two most commonly cited clauses of Chapter 1 are as follows.
- 1.12 – “Quality risk management is a systematic process for the assessment, control, communication and review of risks to the quality of the medicinal product. It can be applied both proactively and retrospectively.”
- 1.4(viii) – “A state of control is established and maintained by developing and using effective monitoring and control systems for process performance and product quality.”
Of these, 1.4(viii) occurs frequently as a critical observation. The other clause relates to quality risk management and this is a regulatory hot-topic. Close behind these two clauses is 1.4(xiii) which covers change controls. The top two most commonly cited parts of Annex 1 are as follows.
- A1.18 – “Environmental monitoring techniques”.
- A1.64 – “Precautions to minimize contamination should be taken during all processing stages including the stages before sterilisation.”
The reference to the way that environmental monitoring is conducted matches the critical observation. The second most common Annex 1 finding is in relation to contamination control. This will become an even bigger focal point when the revised Annex 1 is finally issued [4]. In relation to the critical observations, A1.20 – “Investigating environmental monitoring excursions” comes out relatively high. The top two clauses of Annex 15 are as follows.
- A15.10.5 – “For all cleaning processes an assessment should be performed to determine the variable factors which influence cleaning effectiveness and performance…”
- A15.10.15 – “Where manual cleaning of equipment is performed, it is especially important that the effectiveness of the manual process should be confirmed at a justified frequency…”
Both relate to cleaning, the first to the validation of cleaning and the second to the consistency and controls around manual cleaning. Finally, the following three clauses of Chapter 5 were top.
- C5.20 – “A Quality Risk Management process, which includes a potency and toxicological evaluation, should be used to assess and control the cross-contamination risks presented by the products manufactured…”
- C5.19 – “Cross-contamination should be prevented by attention to design of the premises and equipment…”
- C5.18 – “Contamination of a starting material or of a product by another material or product should be prevented…”
Considering these, it is interesting to note that quality risk management again features, linking back to Chapter 1. With the other two, both relate to contamination control, which again links back to one of the main Chapter 1 observations and one of the central updates that will feature in the revised Annex 1.
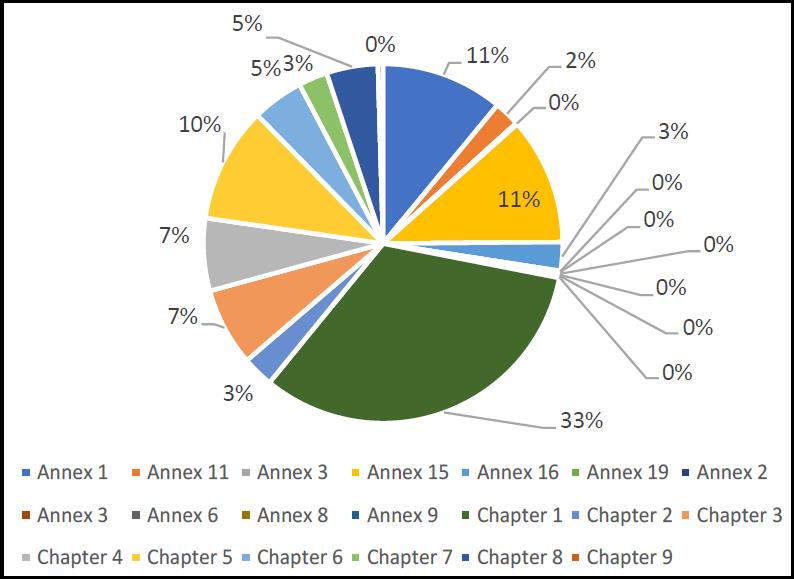
Figure 7. Breakdown of EU GMP major deficiencies according to EU Guidelines to GMP
Summary
The raw data supplied by the MHRA [1] is considerable and countless hours can be spent analysing the trends in ways of greater value to different manufacturers in different locales with different concerns. The aim of this article was to draw out some key themes from the data. Tying the information together, the key trends are deficiencies in the following.
-
Quality risk management.
-
Contamination control strategy, both microbial and cleaning validation.
-
Environmental monitoring.
-
Overall attention to the quality system, especially in relation to change controls and deviations.
-
Appropriate and thorough investigations.
The data also suggests that bigger issues are faced by sterile products manufacturers compared with producers of non-sterile products. While these data are interesting, it must be noted that the data represents only a snapshot in time (given that this is the first time that the MHRA has provided raw data in this format). This limits the assessment of trends and significant shifts – are citations relating to sterile products manufacturers increasing or decreasing? This type of question cannot be answered based on these figures alone. Nevertheless, the data does give some pointers as to the types of areas that GMP
inspectors are either looking more closely into (or at least finding sufficient evidence of non-conformity). Hopefully, the MHRA will produce future data so that longer-term trends can be ascertained. In the meantime, pharmaceutical manufacturers should be assessing their pharmaceutical quality system against the key issues highlighted in this article.
References
[1] Medicines and Healthcare Products Regulatory Agency. MHRA 2018 Deficiency Data. London, UK: MHRA. Available at: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/838906/2018_Deficiency_Data.xlsx (accessed 13 October 2019).
[2] European Medicines Agency. Compilation of Community Procedures on Inspections and Exchange of Information. EMA/572454/2014 Rev 17. London, UK: EMA; October 2014. Available at: https://www.ema.europa.eu/en/documents/regulatoryprocedural-guideline/compilation-community-proceduresinspections-exchange-information_en.pdf (accessed 13 October 2019).
[3] European Commission. Eudralex The Rules Governing Medicinal Products in the European Union, Volume 4 EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use. Annex 1 Manufacture of Sterile Medicinal Products. Brussels, Belgium: European Commission; 2009. Available at: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-4/2008_11_25_gmp-an1_en.pdf (accessed 12 October 2019).
[4] European Medicines Agency. Annex 1 Consultation Document. London, UK: EMA; December 2017. Available at: https://picscheme.org/layout/document.php?id=1268 (accessed 22 December 2017).
Do you have any questions or suggestions? Please contact us at: redaktion@gmp-verlag.de





 | Named User Licence | 12M")

