
Data Integrity in the Quality Control Laboratory
5 min. reading time | by Markus Veit
Published in LOGFILE 12/2021
Introduction
This chapter deals with the practical aspects of implementing the European requirements for data integrity.
A HPLC laboratory for pharmaceutical quality control is used as an example in which data for the batch release of a finished medicinal product is generated. A schematic representation of the underlying process is shown in Figure 1.
Figure 1 Quality control and batch release
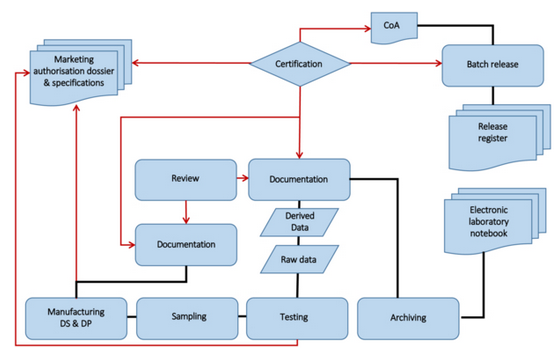
What types of data (in accordance with the WHO guidance) are collected in a chromatography laboratory? The most important types are listed below:
- data from the tested batch and data on personnel who carry out and control the testing process
- sampling and sample storage data, records and observations
- weighing and sample preparation, standards and reagents used
- qualification and calibration data for the pipettes and balances used
- qualification data for all of the devices used
- instrument control data (detector wavelength range, flow, temperature, etc.)
- sequence data in full
- data to be recorded (e. g. data rate, integration parameters, etc.)
- chromatography testing data (initial electronic data, peak areas)
- processed data from chromatography testing (processed electronic data)
- measuring process and device-specific calibrations
- device-specific calculations
- peak areas after integration
- HPLC calibration data
- calculation data (software-based or completed manually)
- trend analyses
- all system suitability test results
- reports generated from electronic data (sample-list printouts, chromatograms, etc.)
- audit trail data and all deviations and changes
- documented observations
- if applicable, calculations carried out using external software (LIMS, Excel) = derived data, results (reportable result), evaluation (with OOS, OOE, OOT)
All of this data should comply with the ALCOA principles – and, in an ideal situation, the ALCOA plus principles.
Figure 2 ALCOA principles of data integrity

A number of these requirements are already contained in the EU GMP Guidelines, i.e. they were introduced before the recent publication of data integrity specifications.
Key aspects of data integrity during the generation of data are examined below. It will be shown that the definition of data is of major importance for a project. The difference between raw data and metadata is not discussed here because the differentiation is difficult. The general term data is used instead. This approach is also taken by the FDA.
Non-compliance with data integrity requirements in analytical laboratories
A number of conflict situations that often arise in relation to data integrity requirements are examined below.
- Access to the key functions of the control and evaluation software (e.g. switching off the audit trail, changing the system time) must be restricted, e.g. limited to IT personnel.
- Access to control and evaluation software must be based on individual login accounts. Group access or anonymous logins should not be possible. User privileges should be limited to the individual job profile.
- A complete qualification and validation of all computers and the control and evaluation systems is absolutely essential.
- A review of audit trails should be carried out before data-based decisions are made. This must be completed before the project is concluded and/or the collected data is used for batch release. Actions must be defined and/or put in place for deviations so that an appropriate investigation can be initiated and completed.
- Test runs and the generation of data for testing purposes are not permitted during testing prior to batch or raw material release if these processes are not carried out in accordance with defined protocols during qualification, validation or the system suitability test.
- There has to be a reason for every subsequent modification, and the reason must be completely and transparently documented. This applies in particular to reintegration and generally to manual integration and changes to manual data entries such as calculation factors, weights and quantities.
- There has to be a reason for every repeated analysis, and the reason must be completely and transparently documented. This type of situation is limited to the investigation of OOS, OOE and OOT results as well as deviations, e.g. failed injections. Repeats can also be indicated when a root cause analysis is carried out. For all repeats, prospectively defined processes must be in place and documentation must be mandatory
- If data lacks robustness and accuracy, e.g. due to an incomplete, unsuitable or old validation or calibration, it may not be used. This can be the case when response factors are used that represent device-specific values, but have not been established as such, or when response factors are not checked and corrected when a device is replaced.
Do you have any questions or suggestions? Please contact us at: redaktion@gmp-verlag.de








 | Named User License | 12M")

